获取国外法森拉药品价格,使用等信息,最快 24 小时回馈
法森拉
什么是Fasenra?
Fasenra(benralizumab)是一种单克隆抗体,会影响人体免疫系统的作用。贝拉珠单抗的作用是降低嗜酸性粒细胞的水平,嗜酸性粒细胞是一种可能导致哮喘症状的白细胞。
Fasenra注射剂是一种处方药,与其他哮喘药物一起使用可帮助控制至少12岁的成人和儿童的严重哮喘。
Fasenra适用于哮喘患者无法通过其他药物控制的情况。
重要信息
在收到Fasenra之前,请告知医生您的所有医疗状况或过敏,所用的所有药物以及您是孕妇还是母乳喂养。
在服药之前
如果您对贝那利珠单抗过敏,则不应使用Fasenra进行治疗。
告诉医生您是否曾经:
寄生虫感染(例如round虫或tape虫);要么
如果您使用口服或吸入类固醇药物。
如果您怀孕,请按照医生的指示接受此药。怀孕期间不治疗哮喘会危害母亲和婴儿。告诉医生您是否怀孕或计划怀孕。
在使用这种药物时母乳喂养婴儿可能并不安全。向您的医生询问任何风险。
不应将Fasenra应用于12岁以下的孩子。
Fasenra如何服用?
在开始使用Fasenra治疗之前,您的医生可能会进行测试以测量您的嗜酸性粒细胞水平。
前3剂每4周一次将Fasenra皮下注射(皮下注射),然后每8周一次。
Fasenra装有单剂量预填充注射器和单剂量自动注射器。
医护人员将使用单剂量预填充注射器注射Fasenra。
如果您的医疗保健提供者决定您或护理人员可以注射Fasenra,则您或您的护理人员应接受使用Fasenra Pen自动注射器进行正确准备和注射的培训。
请非常仔细地遵循医生的剂量说明。
如果您一直在使用类固醇药物,除非您的医生告诉您,否则不要停止使用它。
Fasenra并非哮喘发作的急救药物。仅使用速效吸入药来发作。如果您的速效药物无效,请就医。
您可能需要经常进行医学检查,以帮助您的医生确定用Fasenra治疗您的时间。
哮喘经常用药物联合治疗。按照指示使用所有药物,并阅读所有药物指南。未经医生建议,请勿更改剂量或用药时间表。由于手术,疾病,压力或最近的哮喘发作,您的剂量需求可能会改变。告诉医生您是否有任何药物停止工作。
Fasenra剂量信息
成年人哮喘的常规剂量:
前3剂,每4周一次皮下注射30 mg,此后每8周一次皮下注射
评论:
-该药物应注射到上臂(仅用于护理人员),大腿或腹部。
用途:用于患有嗜酸性表型的重度哮喘患者的附加维持治疗
哮喘的常规儿科剂量:
12岁以上:
前3剂,每4周一次皮下注射30 mg,此后每8周一次皮下注射
评论:
-该药物应注射到上臂(仅用于护理人员),大腿或腹部。
用途:用于对12岁及以上患有嗜酸性表型的严重哮喘患者进行附加维持治疗
如果我错过剂量怎么办?
如果您错过了Fasenra注射预约,请致电您的医生以获取指导。
如果我服药过量怎么办?
由于这种药物是由医疗专业人员在医疗环境中服用的,因此服用过量的可能性不大。
接收Fasenra时应该避免什么?
关于食物,饮料或活动的任何限制,请遵循医生的指示。
Fasenra副作用
如果您对Fasenra有过敏反应迹象,请寻求紧急医疗救助:荨麻疹,皮疹;呼吸困难,头晕;脸,嘴唇,舌头或喉咙肿胀。
如果您有以下情况,请立即致电您的医生:
新的或恶化的哮喘症状。
常见的Fasenra副作用可能包括:
咽喉痛;要么
头痛。
这不是副作用的完整列表,并且可能会发生其他副作用。打电话给您的医生,征求有关副作用的医疗建议。您可以通过1-800-FDA-1088向FDA报告副作用。
还有哪些其他药物会影响Fasenra?
其他药物可能与贝那利珠单抗相互作用,包括处方药和非处方药,维生素和草药产品。告诉您的医生您目前所有的药物以及您开始或停止使用的任何药物。
版权所有1996-2020 Cerner Multum,Inc.版本:1.01。
注意:本文档包含有关贝拉珠单抗的副作用信息。此页面上列出的某些剂型可能不适用于Fasenra品牌。
对于消费者
适用于贝那利珠单抗:皮下溶液
需要立即就医的副作用
除了其所需的作用,贝那利珠单抗(Fasenra中包含的活性成分)可能会引起一些不良作用。尽管并非所有这些副作用都可能发生,但如果确实发生了,则可能需要医疗护理。
服用贝那利珠单抗时,如果发生以下任何副作用,请立即与医生联系:
不常见
- 快速的心跳
- 发热
- 荨麻疹,瘙痒或皮疹
- 嘶哑
- 刺激
- 关节疼痛,僵硬或肿胀
- 皮肤发红
- 眼睑,脸,嘴唇,手或脚肿胀
- 胸闷
- 呼吸困难或吞咽
发病率未知
- 咳嗽
- 头晕
- 眼睑或眼睛,面部,嘴唇或舌头周围浮肿或肿胀
- 异常疲倦或虚弱
不需要立即就医的副作用
贝那珠单抗可能会发生一些副作用,通常不需要医治。随着身体对药物的适应,这些副作用可能会在治疗期间消失。另外,您的医疗保健专业人员可能会告诉您一些预防或减少这些副作用的方法。
请咨询您的医疗保健专业人员,是否持续存在以下不良反应或令人讨厌,或者是否对这些副作用有任何疑问:
比较普遍;普遍上
- 头痛
不常见
- 出血,起泡,灼热,寒冷,皮肤变色,压力感,荨麻疹,感染,炎症,瘙痒,肿块,麻木,疼痛,皮疹,发红,疤痕,酸痛,刺痛,肿胀,压痛,刺痛,溃疡或注射部位的温度
- 身体疼痛或疼痛
- 拥塞
- 喉咙干燥或酸痛
- 流鼻涕
- 脖子上的腺体肿胀
- 声音变化
对于医疗保健专业人员
适用于贝那利珠单抗:皮下溶液
过敏症
未报告频率:过敏反应,血管性水肿[参考]
免疫学的
未报告频率:免疫原性[参考]
本地
常见(1%至10%):注射部位反应(例如疼痛,红斑,瘙痒,丘疹) [Ref]
呼吸道
常见(1%至10%):咽炎(细菌性,病毒性或链球菌性),咳嗽[参考]
其他
常见(1%至10%):发热[参考]
神经系统
常见(1%至10%):头痛[参考]
皮肤科
常见(1%至10%):荨麻疹,皮疹[参考]
参考文献
1.“产品信息。Fasenra(贝拉珠单抗)。”阿斯特拉-塞内卡制药公司,威明顿,DE。
某些副作用可能没有报道。您可以将其报告给FDA。
说明FASENRA药注射器的管理(医疗保健提供商)
请参阅图1以确定要在给药步骤中使用的预填充注射器组件。
图1
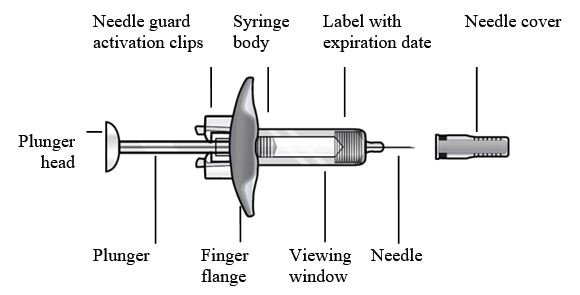
请勿触摸护针器激活夹,以防过早激活护针器。
1个 | 握住注射器主体而不是柱塞,以从托盘中取出预装的注射器。检查注射器上的失效日期。注射器中可能包含小气泡;这很正常。给药前请勿排出气泡。 | |
2 | 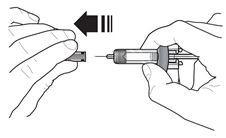 | 准备注射之前,请勿取下针头盖。握住注射器主体,并笔直拉出以除去针头盖。卸下针头盖时,请勿握住柱塞或柱塞头,否则柱塞可能会移动。如果预填充的注射器损坏或污染(例如,未安装针头盖而掉落),请丢弃并使用新的预填充的注射器。 |
3 | 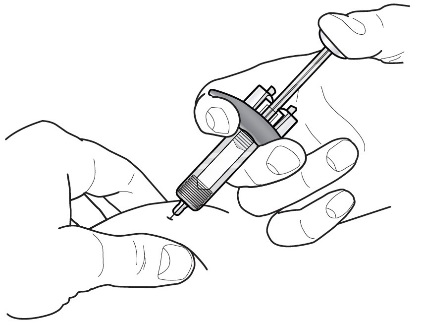 | 轻轻捏住皮肤,然后将针头插入推荐的注射部位(即上臂,大腿或腹部)。 |
4 | 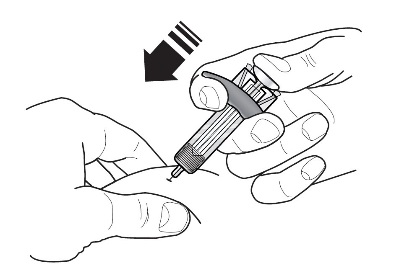 | 完全推入柱塞,直到柱塞头完全位于护针器激活夹之间,以注入所有药物。这是激活护针器所必需的。 |
5 | 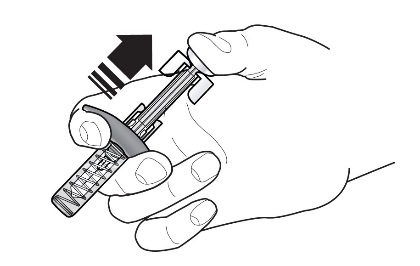 | 注射后,保持柱塞头上的压力并从皮肤上拔下针头。释放柱塞头上的压力,以使护针器盖住针头。不要重新盖好预填充的注射器。 |
6 | 将用过的注射器丢入锐器容器中。 | |
2.4说明FASENRA PEN的管理
请参阅FASENRA PEN“使用说明”对FASENRA PEN的准备和管理更详细的说明[见使用说明。在医疗保健提供者确定合适的情况下,患者可以自行注射或患者护理人员可以皮下注射FASENRA PEN 。
Fasenra的适应症和用法
Fasenra适用于12岁及以上重症哮喘伴嗜酸性表型的患者的附加维持治疗[见临床研究(14) ] 。
使用限制:
- •
- Fasenra不适合治疗其他嗜酸性疾病。
- •
- Fasenra不适用于缓解急性支气管痉挛或哮喘状态。
Fasenra剂量和管理
推荐剂量
Fasenra仅用于皮下使用。
Fasenra的推荐剂量为前3剂,每4周一次,每次30 mg,然后每8周一次,通过皮下注射至上臂,大腿或腹部。
一般管理说明
Fasenra旨在在医疗保健提供者的指导下使用。根据临床实践,建议在施用生物制剂后对患者进行监测[见警告和注意事项(5.1) ] 。
将Fasenra放入大腿或腹部。如果医护人员或护理人员进行注射,也可以使用上臂。给药前,通过将纸箱在室温下放置约30分钟来加热Fasenra。给药前目视检查Fasenra是否存在颗粒物和变色。 Fasenra透明至乳白色,无色至浅黄色,可能包含一些半透明或白色至灰白色颗粒。如果液体混浊,变色或包含大颗粒或异物,请勿使用Fasenra。
预装注射器
预填充的注射器由医疗保健提供者管理。
自动注射器(Fasenra PEN™)
Fasenra PEN适用于患者/护理人员。患者/护理人员可以在接受皮下注射技术的适当培训后,以及在医疗保健提供者确定合适的情况下进行注射。
Fasenra预充式注射器的管理说明(医疗保健提供者)
请参阅图1以确定要在给药步骤中使用的预填充注射器组件。
图1

请勿触摸护针器激活夹,以防过早激活护针器。
1个 | 握住注射器主体而不是柱塞,以从托盘中取出预装的注射器。检查注射器上的失效日期。注射器中可能包含小气泡;这很正常。给药前请勿排出气泡。 | |
2 |  | 准备注射之前,请勿取下针头盖。握住注射器主体,并笔直拉出以除去针头盖。卸下针头盖时,请勿握住柱塞或柱塞头,否则柱塞可能会移动。如果预填充的注射器损坏或污染(例如,未安装针头盖而掉落),请丢弃并使用新的预填充的注射器。 |
3 |  | 轻轻捏住皮肤,然后将针头插入推荐的注射部位(即上臂,大腿或腹部)。 |
4 |  | 完全推入柱塞,直到柱塞头完全位于护针器激活夹之间,以注入所有药物。这是激活护针器所必需的。 |
5 |  | 注射后,保持柱塞头上的压力并从皮肤上拔下针头。释放柱塞头上的压力,以使护针器盖住针头。不要重新盖好预填充的注射器。 |
6 | 将用过的注射器丢入锐器容器中。 | |
2.4 Fasenra PEN的管理说明
有关Fasenra PEN的制备和管理的更多详细说明,请参阅Fasenra PEN的“使用说明” [请参阅使用说明]。在医疗保健提供者确定适当的情况下,患者可以自行注射或患者护理人员可以皮下注射Fasenra PEN 。
剂型和优势
Fasenra为澄清至乳白色,无色至浅黄色溶液,可能包含一些半透明或白色至灰白色颗粒。
- •
- 注射:在单剂量预装注射器中的30 mg / mL溶液。
- •
- 注射:在单剂量自动注射Fasenra PEN中的30 mg / mL溶液。
禁忌症
Fasenra是对贝那利珠单抗或其任何赋形剂过敏的患者的禁忌症[参见警告和注意事项(5.1) ] 。
警告和注意事项
过敏反应
服用Fasenra后发生超敏反应(例如,过敏反应,血管性水肿,荨麻疹,皮疹)。这些反应通常在给药后数小时内发生,但在某些情况下会延迟发作(即几天)。如果发生超敏反应,应停止使用Fasenra [见禁忌症(4) ] 。
急性哮喘症状或疾病恶化
Fasenra不应用于治疗急性哮喘症状或急性加重。不要使用Fasenra来治疗急性支气管痉挛或哮喘状态。如果开始使用Fasenra治疗后哮喘仍未得到控制或恶化,患者应寻求医疗建议。
减少皮质类固醇剂量
开始使用Fasenra进行治疗时,请勿突然终止全身或吸入皮质类固醇激素治疗。如果合适,应逐步减少皮质类固醇激素的剂量,并在医生的直接监督下进行。皮质类固醇剂量的减少可能与全身性戒断症状和/或先前被全身性皮质类固醇疗法抑制的暴露状况有关。
寄生虫(蠕虫)感染
嗜酸性粒细胞可能参与了对某些蠕虫感染的免疫反应。具有已知蠕虫感染的患者被排除在临床试验之外。尚不知道Fasenra是否会影响患者对蠕虫感染的反应。
在开始使用Fasenra进行治疗之前,请对已存在蠕虫感染的患者进行治疗。如果患者在接受Fasenra治疗时被感染并且对抗蠕虫药治疗无反应,请停止使用Fasenra治疗,直到感染消退。
不良反应
其他部分将详细描述以下不良反应:
- •
- 过敏反应[请参阅警告和注意事项(5.1) ]
临床试验经验
由于临床试验是在广泛不同的条件下进行的,因此无法将在某种药物的临床试验中观察到的不良反应率直接与另一种药物在临床试验中观察到的不良反应率进行比较,并且可能无法反映实际中观察到的不良反应率。
在第1、2和3个试验中,有1,808例患者接受了至少1剂Fasenra的剂量[见临床研究(14) ] 。下述数据反映了1,663例患者的Fasenra暴露情况,包括1,556例至少暴露24周和1,387例至少暴露48周。 Fasenra的安全暴露来自两项48期疗程的三期安慰剂对照研究(试验1和2)[Fasenra每4周(n = 841),Fasenra每4周服用3剂,然后每8周(n = 822)和安慰剂(n = 847)]。虽然临床试验中包括每4周一次Fasenra的给药方案,但Fasenra每4周一次给药3剂,此后每8周一次为推荐剂量[参见剂量和给药方法(2.1) ] 。研究的人口年龄在12至75岁之间,其中64%为女性,而79%为白人。
表1中显示了发生率大于或等于3%的不良反应。
| ||
不良反应 | 法森拉 (N = 822) % | 安慰剂 (N = 847) % |
头痛 | 8 | 6 |
发热 | 3 | 2 |
咽炎* | 5 | 3 |
过敏反应† | 3 | 3 |
28周试用
试验3接受Fasenra(n = 73)或安慰剂(n = 75)治疗28周后的不良反应包括头痛(分别为8.2%和5.3%)和发烧(在Fasenra中的发生率比安慰剂高)。分别为2.7%和1.3%) [参见临床研究(14) ] 。 Fasenra的其余不良反应发生频率与安慰剂相似。
注射部位反应
在试验1和2中,用Fasenra治疗的患者发生注射部位反应(例如,疼痛,红斑,瘙痒,丘疹)的发生率为2.2%,而使用安慰剂治疗的患者发生率为1.9%。
免疫原性
与所有治疗性蛋白质一样,具有免疫原性的潜力。抗体形成的检测高度依赖于测定的灵敏度和特异性。另外,在测定中观察到的抗体(包括中和抗体)阳性的发生率可能受到几个因素的影响,这些因素包括测定方法,样品处理,样品收集的时机,伴随用药和基础疾病。由于这些原因,将下述研究中的贝那利珠单抗抗体的发生率与其他研究或其他产品中抗体的发生率进行比较可能会产生误导。
总体而言,在48到56周的治疗期间内,按推荐的剂量方案用Fasenra治疗的患者中有13%出现了治疗性抗药抗体反应。用Fasenra治疗的患者中,共有12%产生了中和抗体。与抗体阴性患者相比,抗药物抗体滴度高的患者中,抗贝拉珠单抗抗体与贝拉珠单抗的清除率增加和血液嗜酸性粒细胞水平升高有关。没有观察到抗药物抗体与疗效或安全性相关的证据。
数据反映了在特定测定中测试结果对贝拉珠单抗抗体呈阳性的患者百分比。
上市后经验
除了临床试验中报告的不良反应外,在批准使用Fasenra的过程中还发现了以下不良反应。由于这些反应是从不确定大小的人群中自愿报告的,因此并非总是能够可靠地估计其发生频率或建立与药物暴露的因果关系。由于事件的严重性,报告频率,与Fasenra的因果关系或这些因素的组合,因此选择将这些事件包括在内。
免疫系统疾病:过敏反应,包括过敏反应。
药物相互作用
尚未进行正式的药物相互作用研究。
在特定人群中的使用
怀孕
怀孕暴露登记
有一个怀孕暴露注册表可以监视怀孕期间暴露于Fasenra的妇女的怀孕结局。医疗保健提供者可以致电1-877-311-8972或访问mothertobaby.org/Fasenra来招募患者或鼓励患者参加。
风险摘要
临床试验中怀孕暴露的数据不足以告知与药物相关的风险。妊娠晚期,单克隆抗体如贝那利珠单抗在胎盘中转运。因此,在妊娠晚期,对胎儿的潜在影响可能更大。在食蟹猴中进行的产前和产后发育研究中,没有证据表明在整个妊娠期静脉内注射贝那利珠单抗对胎儿产生伤害,其剂量所产生的暴露量约为最大推荐人类剂量(MRHD)30毫克时的暴露量的310倍。 SC [请参见数据]。
在美国普通人群中,临床公认的怀孕中主要先天缺陷和流产的估计背景风险分别为2%至4%和15%至20%。
临床注意事项
与疾病相关的孕产妇和/或胚胎/胎儿风险:
有证据表明,在哮喘控制不良或中度的妇女中,母亲先兆子痫的风险增加,而早产,低出生体重和新生儿的胎龄较小。应密切监测孕妇的哮喘控制水平,并根据需要调整治疗以维持最佳控制。
数据
动物资料
在一项产前和产后发育研究中,怀孕的食蟹猕猴从GD20开始至GD22(取决于妊娠确定),从GD35开始,在整个妊娠期之后每14天一次和产后1个月(最多14剂)接受贝那利珠单抗。产生的暴露量约为MRHD的310倍(以AUC为基础,母体IV剂量每2周一次达到30 mg / kg)。直至出生后6.5个月,贝那珠单抗均未对胎儿或新生儿生长(包括免疫功能)产生不良影响。没有证据表明与治疗有关的外部,内脏或骨骼畸形。贝那珠单抗在食蟹猴中没有致畸作用。贝拉珠单抗在食蟹猴中穿过胎盘。产后第7天,母亲和婴儿的Benralizumab浓度大致相等,但后来的婴儿较低。产后6个月,幼猴的嗜酸性粒细胞计数被抑制,并逐渐恢复。然而,在此期间,未观察到一只幼猴的嗜酸性粒细胞计数恢复。
哺乳期
风险摘要
没有关于人乳或动物乳中存在贝那珠单抗的信息,并且尚不知道贝那珠单抗对母乳喂养婴儿和产奶量的影响。但是,贝那利珠单抗是人源化的单克隆抗体(IgG1 /κ类),人乳中存在少量的免疫球蛋白G(IgG)。如果将贝拉利珠单抗转移到人乳中,则在胃肠道局部暴露和婴儿对贝拉珠单抗的潜在有限全身性暴露的影响尚不清楚。应当考虑母乳喂养对发育和健康的益处,以及母亲对贝拉珠单抗的临床需求以及贝拉珠单抗或潜在的母体状况对母乳喂养的孩子的任何潜在不利影响。
儿科用
在3期急性加重试验中,有108名12至17岁的青少年患有哮喘(试验1:n = 53,试验2:n = 55)。其中46例接受安慰剂,40例每4周接受3剂Fasenra,其后每8周接受一次,22例每4周接受Fasenra。患者被要求具有的2次或更多基线需要在过去12个月口服或全身皮质类固醇治疗和肺功能下降哮喘发作史(预支气管扩张FEV 1 <90%),尽管具有中等或高剂量ICS和正规治疗有或没有OCS或其他控制疗法的LABA。根据人群药代动力学分析,贝那利珠单抗在12至17岁的青少年中的药代动力学与成年人一致,并且血液嗜酸性粒细胞计数的减少与在同样的Fasenra治疗后的成年人中观察到的相似。青少年的不良事件概况通常与3期研究中的总体人群相似[见不良反应(6.1) ] 。尚未确定12岁以下患者的安全性和有效性。
老人用
在贝那利珠单抗临床试验的患者总数中,13%(n = 320)为65岁及以上,而0.4%(n = 9)为75岁及以上。在这些患者和年轻患者之间未观察到安全性或有效性的总体差异,其他报告的临床经验也未发现老年患者和年轻患者之间反应的差异,但是不能排除某些老年患者的敏感性更高。
过量
在临床试验中,对嗜酸性粒细胞疾病患者的皮下给药剂量最高为200 mg,而没有剂量相关毒性的证据。
对于贝那利珠单抗过量,尚无特效治疗方法。如果发生用药过量,应在必要的情况下通过适当的监测对患者进行支持治疗。
Fasenra说明
Benralizumab是对白介素5受体α亚基(IL-5Rα)有选择性的人源化单克隆抗体(IgG1 /κ-class)。 Benralizumab是通过重组DNA技术在中国仓鼠卵巢细胞中产生的。贝那珠单抗的分子量约为150 kDa。
Fasenra(benralizumab)注射液是无菌,不含防腐剂,透明至乳白色,无色至浅黄色的溶液,适合皮下注射。由于贝那利珠单抗是一种蛋白质,因此溶液中可能存在一些半透明或白色至灰白色的颗粒。每个单剂量预填充注射器或单剂量自动注射器可递送1 mL,其中包含30 mg贝那利珠单抗,L-组氨酸(1.4 mg); L-组氨酸盐酸盐一水合物(2.3 mg);聚山梨酯20(0.06毫克); α,α-海藻糖二水合物(95毫克);和USP注射用水。单剂量预填充注射器或单剂量自动注射器包含一个1毫升玻璃注射器,该注射器带有29号½英寸长柄不锈钢针。
Fasenra-临床药理学
作用机理
Benralizumab是一种人源化岩藻糖基化单克隆抗体(IgG1,kappa),它以11 pM的解离常数直接结合人白介素5受体(IL-5Rα)的α亚基。 IL-5受体在嗜酸性粒细胞和嗜碱性粒细胞的表面表达。在体外环境中,贝那利珠单抗的Fc域中不存在岩藻糖可促进与免疫效应细胞(如自然杀伤(NK)细胞)上的FcɣRIII受体的结合(45.5 nM),从而导致嗜酸性粒细胞和嗜碱性粒细胞通过抗体依赖性凋亡细胞介导的细胞毒性(ADCC)。
炎症是哮喘发病机理中的重要组成部分。炎症涉及多种细胞类型(例如肥大细胞,嗜酸性粒细胞,嗜中性粒细胞,巨噬细胞,淋巴细胞)和介体(例如组胺,类花生酸,白三烯,细胞因子)。 Benralizumab通过结合IL-5Rα链,通过ADCC减少嗜酸性粒细胞。然而,贝那利珠单抗在哮喘中的作用机制尚未确定。
药效学
在52周的2期剂量范围试验中,哮喘患者接受了3剂贝那利珠单抗[2 mg(n = 81),20 mg(n = 81)或100 mg(n = 222)]或安慰剂( n = 222)。在前3剂中,每4周施用一次所有剂量,然后在其后每8周中施用一次。在2 mg,20 mg和100 mg贝那利珠单抗组和安慰剂组中,基线时血液中嗜酸性粒细胞水平的中位数分别为310、280、190和190个细胞/μL。观察到血液中嗜酸性粒细胞的剂量依赖性减少。在最后一次给药时(第40周),在2 mg,20 mg和100 mg贝那利珠单抗组和安慰剂组中,血液嗜酸性粒细胞计数分别为100、50、40、170细胞/μL。
在一项2期试验中,给药后24小时观察到血液嗜酸性粒细胞计数减少。
在试验1和2中,在以推荐剂量进行贝那利珠单抗的SC给药后,血嗜酸性粒细胞减少至中位数绝对血嗜酸性粒细胞计数为0细胞/μL [参见临床研究(14) ] 。在治疗的第一个观察到的时间点(即4周)可以看到这种减少幅度,并且在整个治疗期间一直保持这种减少幅度。
贝那利珠单抗治疗还与血嗜碱性粒细胞减少有关,这在所有临床研究中均得到一致观察。在2期剂量范围试验中,通过流式细胞仪测量了嗜碱性粒细胞计数。在2 mg,20 mg和100 mg贝那利珠单抗组和安慰剂组中,血液嗜碱性粒细胞计数的中位数分别为45、52、46和40个细胞/ µL。在52周时(最后一剂之后12周),在2 mg,20 mg和100 mg贝那利珠单抗组和安慰剂组中,血液嗜碱性粒细胞计数分别为42、18、17和46细胞/ µL。
药代动力学
在哮喘患者中,皮下给药剂量范围为20至200 mg,贝那利珠单抗的药代动力学与剂量成比例。
吸收性
皮下给予哮喘患者后,吸收半衰期约为3.5天。根据人群药代动力学分析,估计的绝对生物利用度约为59%,并且在腹部,大腿或手臂给药中,相对生物利用度没有临床相关的差异。
分配:
根据人群药代动力学分析,对于70公斤的个体,贝那利珠单抗的中心分布和外周分布分别为3.1 L和2.5L。
代谢:
Benralizumab是一种人源化IgG1单克隆抗体,可被广泛分布在体内且不限于肝组织的蛋白水解酶降解。
消除:
从人群药代动力学分析来看,贝那利珠单抗显示出线性药代动力学,并且没有靶受体介导的清除途径的证据。对于体重70公斤的受试者,贝那利珠单抗的估计典型全身清除率(CL)为0.29 L / d。皮下给药后,消除半衰期约为15.5天。
特定人群:
年龄:
根据人群药代动力学分析,年龄不影响贝那利珠单抗清除率。
性别,种族:
人群药代动力学分析表明,性别和种族对贝那利珠单抗清除率没有显着影响。
肾功能不全:
尚未进行正式的临床研究来研究肾功能不全对贝那利珠单抗的影响。根据人群药代动力学分析,在肌酐清除率介于30和80 mL / min之间的受试者和肾功能正常的患者中,贝那利珠单抗清除率具有可比性。肌酐清除率值低于30 mL / min的受试者的可用数据有限。但是,贝那利珠单抗不能通过肾脏清除。
肝功能不全:
尚未进行正式的临床研究来研究肝功能损害对贝那利珠单抗的影响。 IgG单克隆抗体并未首先通过肝途径清除;肝功能的变化预计不会影响贝那利珠单抗的清除率。根据人群药代动力学分析,基线肝功能生物标志物(ALT,AST和胆红素)对贝那利珠单抗清除率无临床相关影响。
药物相互作用:
尚未进行正式的药物相互作用研究。
细胞色素P450酶,外排泵和蛋白质结合机制不参与贝那利珠单抗的清除。没有证据表明肝细胞中IL-5Rα的表达,嗜酸性粒细胞的消耗不会产生促炎性细胞因子的慢性全身性改变。
预计贝那利珠单抗对联合用药的药代动力学没有影响。根据人群分析,常用的共同用药对哮喘患者的贝那利珠单抗清除率没有影响。
非临床毒理学
致癌,诱变,生育力受损
尚未进行长期动物研究来评估贝那利珠单抗的致癌潜力。使用动物模型发表的文献表明,IL-5和嗜酸性粒细胞是肿瘤发生部位早期炎症反应的一部分,可以促进肿瘤排斥。然而,其他报道表明嗜酸性粒细胞浸润进入肿瘤可促进肿瘤生长。因此,未知与贝拉利珠单抗等IL-5Rα结合的抗体对人的恶性风险。
男性和女性的生育力均未受到影响,这是因为在接受贝那利珠单抗治疗的9个月食蟹猴的生殖器官中,无不良组织病理学发现,静脉注射剂量最高为25 mg / kg,SC剂量最高为30 mg / kg,每2周一次(以AUC为基础,约为MRHD的400倍和270倍)。
临床研究
Fasenra的哮喘发展计划包括一项52周剂量范围加重试验(NCT01238861),三项确证性试验(试验1 [NCT01928771],试验2 [NCT01914757],试验3 [NCT02075255])和一项12周肺功能试验( NCT02322775)。
剂量范围试验
2期随机,双盲,安慰剂对照,为期52周的剂量范围试验纳入了609名18岁以上的哮喘患者。患者每4周皮下注射2毫克贝那珠单抗,20毫克或100毫克或安慰剂治疗3剂,随后每8周治疗一次。主要终点指标是每年的急性发作率和1秒内的呼气量(FEV 1 ),而ACQ-6是主要的次要终点指标。在过去的12个月中,要求患者有2次或更多次哮喘加重病史(但不超过6次加重病史),需要全身性糖皮质激素治疗;筛查期间ACQ-6评分至少为1.5,两次;筛查时早晨肺功能下降[使用中等或大剂量ICS加LABA进行治疗时,[支气管扩张剂前FEV 1低于90%]。患者按嗜酸性状态分层。接受贝那珠单抗2 mg,20 mg和100 mg的患者的年恶化率分别为-12%(80%CI:-52,18),34%(80%CI:6,54),29%(80% CI:10、44),分别与安慰剂相比(比率0.56)。
该试验的结果以及加重率降低的暴露-响应模型均支持随后试验中贝那珠单抗30 mg的评估[参见临床药理学(12.2和12.3) ] 。 Fasenra未被批准以2 mg,20 mg或100 mg的剂量使用,仅应以建议的30 mg剂量进行给药[参见剂量和用法(2.1) ] 。
确认性试验
试验1和试验2是随机,双盲,平行组,安慰剂对照的急性加重试验,分别针对12岁以上的患者以及持续时间为48周和56周的患者。该试验总共对2510名患者进行了随机分组。在过去的12个月中,患者必须有2次或两次以上哮喘发作的病史,需要口服或全身性皮质类固醇激素治疗,筛查时ACQ-6得分为1.5或更高,并且基线时肺功能降低[支气管扩张剂前FEV 1低于80成人中的百分比,青少年中低于90%],尽管定期接受高剂量吸入皮质类固醇(ICS)(试验1)或中,高剂量ICS(试验2)加长效β受体激动剂(LABA)的治疗口服皮质类固醇(OCS)和其他哮喘控制药物。按地理位置,年龄和血液嗜酸性粒细胞计数(≥300细胞/μL或<300细胞/μL)对患者进行分层。前3剂Fasenra每4周给药一次,然后每4或8周给药一次,与安慰剂相比,作为背景治疗的附加药物。
在整个试验过程中,所有受试者均继续进行了背景哮喘治疗。
试验3是一项针对220名哮喘患者的随机,双盲,平行组OCS减少试验。除了定期使用大剂量ICS和LABA(有或没有其他控制器)外,患者还需要每日服用OCS(每天7.5至40 mg)进行治疗。该试验包括一个为期8周的磨合期,在此期间将OCS滴定至最小有效剂量,而不会失去哮喘控制。出于OCS剂量滴定的目的,研究人员根据患者的FEV 1 ,峰值呼气流量,夜间清醒,支气管扩张药的速效使用或需要增加OCS剂量的任何其他症状,对哮喘控制进行评估。在所有治疗组中,基线OCS剂量中位数均相似。要求患者在过去12个月中血中嗜酸性粒细胞计数大于或等于150个细胞/μL,并且至少有一次加重病史。所有3个治疗组的基线OCS基线中值剂量均为10 mg(范围:8至40 mg)(安慰剂,每4周一次Fasenra,前3剂每4周一次Fasenra,然后每8周一次)。
虽然在试验1、2和3中研究了2种给药方案,但建议的给药方案是前4剂,每4周给药30 mg Fasenra,然后每8周给药一次[参见剂量和给药方法(2.1) ] 。
| 总人口 | |||
|---|---|---|---|
试验1 (N = 1204) | 试用2 (N = 1306) | 试用3 (N = 220) | |
平均年龄(岁) | 49 | 49 | 51 |
女(%) | 66 | 62 | 61 |
白色(%) | 73 | 84 | 93 |
哮喘持续时间,中位数(年) | 15 | 16 | 12 |
从未吸烟(%) | 80 | 78 | 79 |
平均基线FEV 1支气管扩张剂前(L) | 1.67 | 1.76 | 1.85 |
平均基线百分比预测FEV 1 | 57 | 58 | 60 |
平均SABA后FEV 1 / FVC(%) | 66 | 65岁 | 62 |
基线平均嗜酸性粒细胞计数(细胞/μL) | 472 | 472 | 575 |
前一年的平均发作次数 | 3 | 3 | 3 |
病情加重
试验1和试验2的主要终点是服用大剂量ICS和LABA的基线血液嗜酸性粒细胞计数大于或等于300细胞/μL的患者的哮喘急性发作率。哮喘加重定义为哮喘恶化,需要使用口服/全身性皮质类固醇至少3天,和/或急诊就诊需要使用口服/全身性皮质类固醇和/或住院。对于维持口服皮质类固醇激素的患者,需要口服皮质类固醇激素的哮喘发作定义为稳定的口服/全身性皮质类固醇激素至少持续3天的暂时增加或单次可注射剂量的皮质类固醇激素的增加。在试验1中,接受Fasenra的患者中有35%经历了哮喘急性发作,而安慰剂组则为51%。在试验2中,接受Fasenra的患者中有40%经历了哮喘急性发作,而安慰剂组则为51% (表3) 。
| 试用版 | 治疗 | 每年恶化 | |||
|---|---|---|---|---|---|
| |||||
率 | 区别 | 比率(95%CI) | |||
所有恶化 | |||||
试验1 | Fasenra † (n = 267) | 0.74 | -0.78 | 0.49(0.37,0.64) | |
安慰剂(n = 267) | 1.52 | - | - | ||
试用2 | Fasenra † (n = 239) | 0.73 | -0.29 | 0.72(0.54,0.95) | |
安慰剂(n = 248) | 1.01 | - | - | ||
需要住院/急诊室就诊的病情加重 | |||||
试验1 | Fasenra † (n = 267) | 0.09 | -0.16 | 0.37(0.20,0.67) | |
安慰剂(n = 267) | 0.25 | - | - | ||
试用2 | Fasenra † (n = 239) | 0.12 | 0.02 | 1.23(0.64,2.35) | |
安慰剂(n = 248) | 0.10 | - | - | ||
需要住院的病情加重 | |||||
试验1 | Fasenra † (n = 267) | 0.07 | -0.07 | 0.48(0.22,1.03) | |
安慰剂(n = 267) | 0.14 | - | - | ||
试用2 | Fasenra † (n = 239) | 0.07 | 0.02 | 1.48(0.65,3.37) | |
安慰剂(n = 248) | 0.05 | - | - | ||
与试验1中的安慰剂相比,接受Fasenra的患者首次发作的时间更长(图2 )。在试验2中也发现了类似的发现。
图2.首次发作时间的Kaplan-Meier累积发生率曲线,试验1

从试验1和2进行的亚组分析确定,既往病情加重病史和基线嗜酸性粒细胞计数较高的患者可作为治疗反应改善的潜在预测指标。不论基线外周嗜酸性粒细胞计数如何,急性发作率均降低。然而,基线血嗜酸性粒细胞计数≥300细胞/μL的患者比计数<300细胞/μL的患者表现出更大的反应。在这两项试验中,在Fasenra随机分组前12个月内有3次或以上加重病史的患者,其病情加重反应的数值要高于先前加重次数较少的患者。
口服糖皮质激素减少
试验3评估了Fasenra对减少维持性口服糖皮质激素的使用的作用。主要终点是在保持哮喘控制的同时在24至28周内从最终OCS剂量基线降低的百分比(请参见试验说明中哮喘控制的定义)。与安慰剂相比,接受Fasenra的患者在维持哮喘控制的同时,每日维持口服糖皮质激素的剂量有更大的减少。接受Fasenra(95%CI:60,88)的患者每日OCS剂量相对于基线的中位数减少百分比为75%,而接受安慰剂的患者(95%CI:0,33)为25%。与接受安慰剂28的患者(37%)相比,接受Fasenra的48位患者(66%)的OCS剂量降低了50%或更高。在第24至28周,平均最终剂量小于或等于5 mg的患者比例对于Fasenra为59%,对于安慰剂为33%(赔率比2.74、95%CI:1.41、5.31)。在研究期间,只有优化的基线OCS剂量为12.5 mg或更少的患者才有资格实现100%的OCS剂量降低。在这些患者中,接受Fasenra的患者为52%(42名中的22名),接受安慰剂的患者为19%(42名中的8名),OCS剂量降低了100%。导致住院和/或急诊就诊的病情加重也被视为次要终点。在这项为期28周的试验中,接受Fasenra的患者发生1次事件,而接受安慰剂的患者发生14次事件(年率分别为0.02和0.32;比率为0.07、95%CI:0.01、0.63)。
肺功能
在试验1、2和3中将平均FEV 1从基线的变化评估为次要终点。与安慰剂相比,Fasera在FEV 1中相对于基线的平均变化随时间的变化持续改善(图3和表4 )。
图3.试验前支气管扩张剂FEV 1 (L)与基线的平均变化

| |
试用版 | 与支气管扩张剂基线FEV 1 (L)的均值变化与安慰剂的差异 |
1个 | 0.159(0.068,0.249) |
2 | 0.116(0.028,0.204) |
3 | 0.112(-0.033,0.258) |
亚组分析还显示,基线血嗜酸性粒细胞计数更高且病情加重病史更频繁的患者,FEV 1改善更大。
Fasenra的临床开发计划还包括对211名轻度至中度哮喘患者进行的为期12周,随机,双盲,安慰剂对照的肺功能试验。每4周用安慰剂或贝那利珠单抗30 mg SC治疗患者3剂。与安慰剂相比,贝那利珠单抗治疗组的肺功能(通过第12周FEV 1的基线变化)得以改善。
患者报告的结果
在试验1、2和3中评估了哮喘控制问卷6(ACQ-6)和12岁及以上的标准哮喘生活质量问卷(AQLQ(S)+12)。两种方法的应答率均定义为在试验1、2和3(分别为48、56和28周)结束时,将阈值的得分提高0.5或更高。在试验1中,对Fasenra的ACQ-6应答率是60%vs安慰剂50%(赔率1.55; 95%CI:1.09,2.19)。在试验2中,Fasenra的ACQ-6应答率是63%vs安慰剂59%(赔率1.16; 95%CI:0.80,1.68)。在试验1中,对Fasenra的AQLQ(S)+12的响应率是57%对49%的安慰剂(赔率比1.42; 95%CI:0.99、2.02),在试验2中,60%的Fasenra对59%的安慰剂(比值比为1.03; 95%CI:0.70,1.51)。在试验3中看到了类似的结果。
供应/存储和处理方式
供应方式
Fasenra(benralizumab)注射液为无菌,无防腐剂,透明至乳白色,无色至浅黄色溶液,可能包含一些半透明或白色至灰白色颗粒,用于皮下注射,以单剂量预装注射器或单剂量形式提供剂量自动注射器。预填充注射器(包括塞子和盖子)和自动注射器不是用天然橡胶乳胶制成的。
Fasenra可作为:
- •
- 单剂量预填充注射器
纸箱包含一个30 mg / mL单剂量预填充注射器:NDC 0310-1730-30
- •
- 单剂量自动注射器Fasenra PEN
纸箱包含一种30 mg / mL单剂量自动注射器:NDC 0310-1830-30
储存和处理
Store refrigerated at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
If needed, the prefilled syringe and autoinjector may be stored at room temperature up to 77°F (25°C) for a maximum of 14 days in the original carton to protect from light. Once removed from the refrigerator and brought to room temperature (up to 77°F [25°C]), the prefilled syringe and autoinjector must be used within 14 days or discarded.
不要冻结。不要摇晃。 Do not expose to heat.
病人咨询信息
Advise the patients and/or caregivers to read the FDA-approved patient labeling (Patient Information and Instructions for Use for Fasenra PEN) before the patient starts using Fasenra and each time the prescription is renewed as there may be new information they need to know.
Provide proper training to patients and/or caregivers on proper subcutaneous injection technique using the Fasenra PEN, including aseptic technique, and the preparation and administration of Fasenra PEN prior to use. Advise patients to follow sharps disposal recommendations [see Instructions for Use].
过敏反应
Inform patients that hypersensitivity reactions (eg, anaphylaxis, angioedema, urticaria, rash) have occurred after administration of Fasenra. These reactions generally occurred within hours of Fasenra administration, but in some instances had a delayed onset (ie, days). Instruct patients to contact their healthcare provider if they experience symptoms of an allergic reaction [see Warnings and Precautions (5.1) ].
Not for Acute Symptoms or Deteriorating Disease
Inform patients that Fasenra does not treat acute asthma symptoms or acute exacerbations. Inform patients to seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with Fasenra [see Warnings and Precautions (5.2) ] .
Reduction of Corticosteroid Dosage
Inform patients to not discontinue systemic or inhaled corticosteroids except under the direct supervision of a physician. Inform patients that reduction in corticosteroid dose may be associated with systemic withdrawal symptoms and/or unmask conditions previously suppressed by systemic corticosteroid therapy [see Warnings and Precauti
药物状态
- 可用性 仅处方
- 怀孕和哺乳 现有风险数据
- CSA时间表* 不是管制药物
- 审批历史 FDA批准2017
美国日本医生

Gregory Aaen MD

Brian Aalbers DO

Glen Scott DO

Cecile Becker MD

Shruti Badhwar DO

中山秀章 教授

村田朗

山内広平

吉井文均 东海大学名誉教授
